
The pharmaceutical landscape is governed by strict regulatory frameworks that focuses the safety and efficacy of medications. In the United States, two primary pathways exist for drug approval: the New Drug Application (NDA) and the Abbreviated New Drug Application (ANDA).
Understanding the distinctions between these processes is crucial for pharmaceutical companies and healthcare professionals. This guide explores the key differences, processes, and requirements of NDAs and ANDAs, providing a comprehensive overview of how new and generic drugs reach the market.
What is an ANDA (Abbreviated New Drug Application)?
An ANDA is a regulatory submission to the FDA for approval of generic drugs that demonstrates bioequivalence to an already-approved brand-name medication, bypassing the need for extensive clinical trials required for new drug development.
The ANDA pathway represents a streamlined regulatory approach that allows generic pharmaceutical manufacturers to bring cost-effective alternatives to market more efficiently. Rather than conducting the comprehensive safety and efficacy studies required for entirely new medications, ANDA applicants focus on proving their generic product performs similarly to the Reference Listed Drug (RLD) in the human body.
This abbreviated process significantly reduces development costs and timelines while maintaining rigorous quality standards. The FDA’s ANDA review process ensures that generic drugs meet the same strict manufacturing, quality, and performance standards as their brand-name counterparts, providing patients with safe and effective treatment options at reduced costs.
What is an NDA (New Drug Application)?
A New Drug Application (NDA) is the comprehensive regulatory submission that pharmaceutical companies must file with the FDA to obtain approval for marketing a new drug product in the United States. The NDA drug approval process represents the culmination of years of research, development, and clinical testing to demonstrate a medication’s safety and effectiveness.
The NDA serves as the definitive document that contains all scientific data, manufacturing information, and proposed labeling for a new pharmaceutical product. This extensive submission enables the FDA to conduct a thorough evaluation of whether the benefits of the new drug outweigh its risks for the intended patient population. The process ensures that only medications meeting the highest standards for safety, efficacy, and quality reach American patients.
Unlike abbreviated applications for generic drugs, NDAs require original clinical trial data generated specifically for the new drug candidate, making this pathway essential for innovative therapies that address unmet medical needs or provide significant therapeutic advances over existing treatments.
Exploring NDA
The NDA pathway represents the most rigorous route to drug approval in the United States, designed specifically for innovative pharmaceutical products that have never before received FDA authorization. This comprehensive regulatory framework requires sponsors to generate original scientific evidence through years of research and development, distinguishing it from abbreviated pathways available for generic medications.
The NDA process involves substantial investment in both time and resources, typically spanning 10-15 years from initial discovery to market approval. This extensive timeline reflects the complexity of developing novel therapeutic approaches and the FDA’s commitment to thorough evaluation of breakthrough treatments. The process encompasses multiple phases of investigation, from laboratory research through large-scale clinical trials, each building upon previous findings to construct a complete scientific case for the new medication’s therapeutic value.
Essential Components of an NDA Submission
An NDA submission is a detailed and multifaceted document that includes several critical components:
- Clinical Trial Data and Results: This section contains the results of all clinical trials conducted on the drug, including data from Phase I, II, and III trials. These trials assess the drug’s pharmacokinetics, pharmacodynamics, efficacy, and safety in human subjects. The data must demonstrate that the drug is effective for its intended use and has an acceptable safety profile.
- Preclinical Studies and Toxicology Reports: Before clinical trials begin, extensive preclinical studies are conducted to evaluate the drug’s safety in animal models. These studies help identify potential toxic effects and determine safe starting doses for human trials. The NDA includes all relevant toxicology data, including studies on carcinogenicity, genotoxicity, and reproductive toxicity.
- Manufacturing Information and Quality Control: The NDA must provide detailed information on the drug’s manufacturing process, including the facilities where the drug is produced, the methods used to ensure consistent product quality, and the procedures for testing the drug’s purity, potency, and stability.
Related Article: What is Pharmacodynamics?
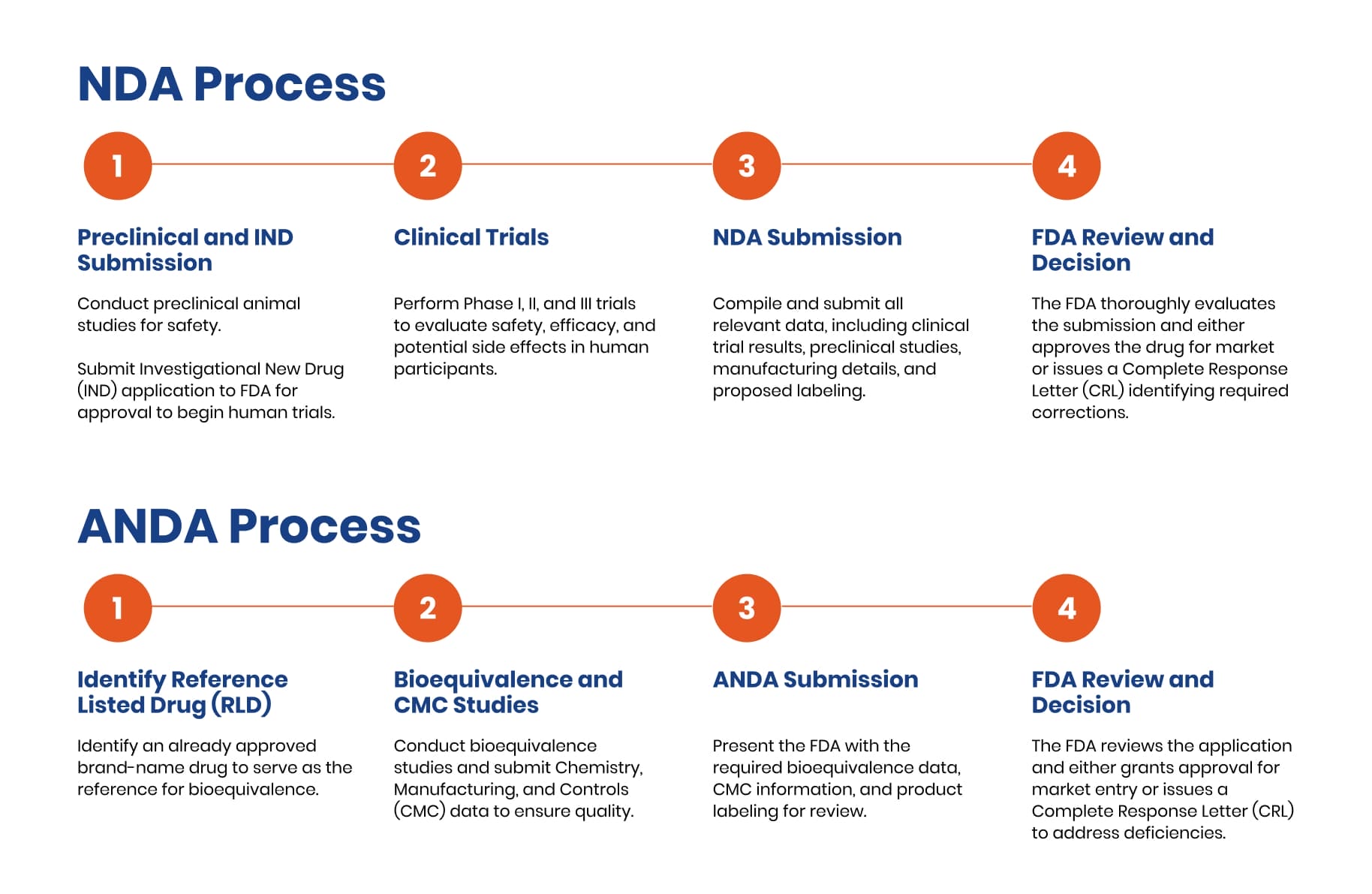
NDA Submission: Process at a Glance
NDA submission is the FDA process where sponsors compile and formally present all required data, studies, and documentation to demonstrate their new drug’s safety and effectiveness for regulatory approval.
- Chemistry, Manufacturing, and Controls (CMC) – Compile comprehensive data on drug substance, drug product specifications, manufacturing processes, and quality control measures.
- Nonclinical Studies – Submit preclinical safety data, including toxicology, pharmacology, and animal studies that support human testing.
- Clinical Trial Documentation – Provide complete Phase I, II, and III clinical trial results demonstrating safety and efficacy in human subjects.
- Labeling Development – Prepare proposed prescribing information, patient labeling, and risk communication materials based on clinical findings.
- Facility and Inspection Readiness – Ensure manufacturing facilities meet FDA standards and are prepared for potential pre-approval inspections.
- User Fee Payment and Administrative Requirements – Submit required FDA user fees and complete all administrative documentation for application processing.
- FDA Review Outcome – Await FDA’s decision through their review process, which may result in approval, Complete Response Letter, or additional data requests.
The NDA Review Process
The FDA review process for an NDA is thorough and multifaceted, involving multiple steps and specialized review teams. The process begins with a preliminary evaluation of the application’s completeness, followed by an in-depth review by the FDA’s multidisciplinary team of experts.
Key steps in the FDA review process include:
- NDA Filing Review: The FDA determines whether the NDA is sufficiently complete to permit a substantive review. If the application is incomplete, the FDA may issue a Refuse to File (RTF) letter outlining the deficiencies.
- Substantive Review: The FDA evaluates the scientific data and information in the NDA, focusing on the drug’s safety, efficacy, and quality. This review may involve consultations with external experts and advisory committees.
- Labeling and Risk Management: The FDA reviews the proposed labeling for accurate reflection of the drug’s benefits and risks. The agency also evaluates any proposed risk management strategies, such as a Risk Evaluation and Mitigation Strategy (REMS), to mitigate potential safety concerns.
- Decision Making: The FDA concludes the review process by making a final decision on the NDA. Possible outcomes include approval, issuance of a Complete Response Letter (CRL) outlining deficiencies that must be addressed, or a request for additional data. In some cases, the FDA may convene an advisory committee to provide recommendations.
How is ANDA Different?
The ANDA pathway revolutionized pharmaceutical accessibility by creating a science-based framework that leverages existing safety and efficacy data from previously approved medications. This regulatory innovation allows generic manufacturers to enter the market without duplicating the extensive and costly research already conducted for brand-name drugs, significantly accelerating the availability of affordable treatment options.
Generic drug development through the ANDA process typically requires 3-5 years compared to the decade-plus timeline for novel drug development. This efficiency stems from the FDA’s recognition that once a drug’s safety and effectiveness have been thoroughly established through the original NDA process, the focus can shift to ensuring manufacturing quality and bioequivalent performance rather than repeating clinical investigations.
The ANDA framework serves a crucial public health function by fostering pharmaceutical competition, which drives down medication costs while maintaining therapeutic standards. Generic drugs approved through this pathway account for approximately 90% of all prescriptions filled in the United States, demonstrating the pathway’s success in balancing regulatory rigor with market accessibility.
Elements of an ANDA Submission
An ANDA submission focuses on several key components that collectively demonstrate the generic drug’s equivalence to the RLD:
- Bioequivalence Studies and Requirements: Bioequivalence studies are the cornerstone of an ANDA submission. These studies compare the pharmacokinetic properties of the generic drug to those of the RLD, specifically measuring parameters such as the rate and extent of absorption. To be considered bioequivalent, the generic drug must fall within an acceptable range of variability compared to the RLD, ensuring no significant differences in safety or efficacy.
- Reference Listed Drug (RLD) Considerations: The ANDA must identify a specific RLD that has already been approved by the FDA. The generic applicant must demonstrate that their product is comparable to this RLD in terms of active ingredients, dosage form, strength, route of administration, and conditions of use.
- Chemistry, Manufacturing, and Controls (CMC) Data: The ANDA must include detailed information on the chemistry, manufacturing, and controls of the generic drug. This includes the formulation and specifications of the drug substance and product, the manufacturing process, quality control measures, and stability data. The CMC data makes sure that the generic product can be consistently produced and meet the same quality standards as the RLD.
The ANDA Review Process
The FDA’s review process for ANDAs is designed to be efficient, reflecting the lower risk associated with generic drugs compared to new, innovative drugs.
The review process begins with a filing review to determine if the application is sufficiently complete. If the application meets the requirements, the FDA proceeds with a detailed assessment of the bioequivalence studies, CMC data development, and other relevant information. The agency may request additional information or clarifications during the review process.
The review timeline for ANDAs is typically shorter than for NDAs, reflecting the reduced need for clinical and preclinical data. The Generic Drug User Fee Amendments (GDUFA) program sets goals for the timely review of ANDAs, with standard reviews generally completed within ten months and priority reviews within eight months.
After completing the review, the FDA may approve the ANDA, allowing the generic product to enter the market. Alternatively, the FDA may issue a Complete Response Letter (CRL) if deficiencies are found. The approval of an ANDA has significant implications for the market, as it introduces competition, typically resulting in lower prices for patients and healthcare systems.

Comparing NDA and ANDA
When comparing NDAs and ANDAs, it is essential to understand the key differences in data requirements, cost and time considerations, and the implications of market exclusivity and patent issues.
Data Requirements and Evidence
NDAs necessitate comprehensive clinical data demonstrating the safety and efficacy of a new drug. This includes extensive clinical trials and preclinical studies.
In contrast, ANDAs focus on bioequivalence data, requiring evidence that the generic product performs similarly to the Reference Listed Drug (RLD) in terms of absorption and effect. This streamlined approach eliminates the need for costly and time-consuming clinical trials, provided the generic meets the bioequivalence criteria.
Cost and Time Considerations
The financial and time investments for NDA and ANDA applicants differ significantly. NDAs involve high costs and long timelines due to the need for extensive research and clinical trials, which can span several years.
ANDAs are less costly and quicker to develop as they bypass the clinical trial phase, focusing solely on demonstrating bioequivalence. This efficiency often results in a faster path to market, allowing generic drugs to be introduced more rapidly.
Market Exclusivity and Patent Issues
NDAs typically offer a period of market exclusivity, granting the innovator company exclusive rights to market the drug for a specified time, usually five years. This exclusivity period can be extended for pediatric studies or new clinical indications.
On the other hand, ANDA applicants may face patent challenges, including potential litigation, if they seek approval before the expiration of the RLD’s patents. Successful ANDA applicants can benefit from 180 days of exclusivity if they are the first to file, allowing them a temporary monopoly before other generics enter the market.
Navigating the Pharmaceutical Approval Maze
The NDA and ANDA processes are fundamental to the pharmaceutical industry. While NDAs are essential for introducing new, innovative therapies, ANDAs provide a pathway for affordable generic alternatives so patients have access to essential medicines.
As advancements in drug development continue, these processes will remain integral to maintaining the balance between innovation and accessibility.
Frequently Asked Questions
Q: What is ANDA?
A: An Abbreviated New Drug Application allows FDA to approve a generic drug by demonstrating bioequivalence to a reference listed drug (RLD). ANDAs rely on existing findings for safety and efficacy, emphasizing sameness and quality rather than repeating full clinical trials.
Q: What is NDA?
A: A New Drug Application is the FDA submission for a new branded medicine, providing full data on quality, safety, and efficacy so FDA can decide if benefits outweigh risks and labeling is appropriate.
Q: What is an NDA drug?
A: An NDA drug is the innovator product that undergoes FDA’s full review of CMC, nonclinical, and clinical evidence. Approval permits U.S. marketing with FDA-approved labeling.
Q: How long does an NDA submission take?
A: Timelines vary with program, data package, inspections, and review designations. Standard NDA reviews follow target review goals; expedited programs may shorten steps when criteria are met.





{kind=link}